Physical chemistry is central to all of molecular science, seeking fundamental understanding in areas as diverse as the structure and function of biomolecules, to molecular electronics, nanomaterials, molecular dynamics and reactivity, and biosensors. In our department, faculty research focuses on developing fundamental techniques, instrumentation, and computational simulation methods, and applying these to biological, materials, and energy problems.
See the descriptions below for outlines of each faculty’s research focus.
Asher group
Physical Chemistry, Biophysical Chemistry, and Materials Science with a touch of Analytical Chemistry
 Research in the Asher group is interdisciplinary and involves the disciplines of Physical Chemistry, Biophysics, Spectroscopy, and Materials Science to learn about the chemistry of our own bodies as well as the chemistry of the world around us. UV Resonance Raman Spectroscopy and Crystalline Colloidal Array Photonic Crystals are the two main areas of our research. Our work in spectroscopy examines the mechanisms of protein folding and protein aggregation to form fibrils that are connected to awful diseases such as Alzheimers and Huntington’s disease. The goal is to understand the mechanisms of fibril assembly and to develop insights into molecular processes that would destabilize these fibrils and prevent their formation. Our work in materials and optics develops photonic crystals to control the propagation of light and to sense volume phase transitions in responsive hydrogels and organogels. We developed sensor technologies to sense glucose in tear fluid to aid people with diabetes mellitus. Our responsive
Research in the Asher group is interdisciplinary and involves the disciplines of Physical Chemistry, Biophysics, Spectroscopy, and Materials Science to learn about the chemistry of our own bodies as well as the chemistry of the world around us. UV Resonance Raman Spectroscopy and Crystalline Colloidal Array Photonic Crystals are the two main areas of our research. Our work in spectroscopy examines the mechanisms of protein folding and protein aggregation to form fibrils that are connected to awful diseases such as Alzheimers and Huntington’s disease. The goal is to understand the mechanisms of fibril assembly and to develop insights into molecular processes that would destabilize these fibrils and prevent their formation. Our work in materials and optics develops photonic crystals to control the propagation of light and to sense volume phase transitions in responsive hydrogels and organogels. We developed sensor technologies to sense glucose in tear fluid to aid people with diabetes mellitus. Our responsive
materials also sense microorganisms that bind to our novel protein hydrogel materials.
Click here to visit the Asher group’s website.
Chong group
Computational Biophysics, Biomolecular Simulations
 The Chong research group has been focused on the development and application of molecular simulation approaches to characterize the pathways, free energy landscapes, and kinetics of a variety of biological processes, including large protein conformational transitions and protein binding. We have also been developing simulation approaches for aiding the design of engineered protein-based conformational switches in collaboration with Stewart Loh (SUNY Upstate Medical University). We are developers of a) an upcoming AMBER force field for biomolecular simulation (AMBER ff15ipq) and b) WESTPA, a freely available, highly scalable software package for the execution and analysis of the weighted ensemble path sampling strategy for the simulation of long-timescale processes that are rare events (e.g., protein folding and protein binding). Figure: Direct observations of shifts in the intermolecular beta-sheet of a protein-peptide complex using atomically detailed molecular dynamics simulations and large-scale computing resources. Results provide detailed views of how nature might correct for 'mistakes' in binding orientation.
The Chong research group has been focused on the development and application of molecular simulation approaches to characterize the pathways, free energy landscapes, and kinetics of a variety of biological processes, including large protein conformational transitions and protein binding. We have also been developing simulation approaches for aiding the design of engineered protein-based conformational switches in collaboration with Stewart Loh (SUNY Upstate Medical University). We are developers of a) an upcoming AMBER force field for biomolecular simulation (AMBER ff15ipq) and b) WESTPA, a freely available, highly scalable software package for the execution and analysis of the weighted ensemble path sampling strategy for the simulation of long-timescale processes that are rare events (e.g., protein folding and protein binding). Figure: Direct observations of shifts in the intermolecular beta-sheet of a protein-peptide complex using atomically detailed molecular dynamics simulations and large-scale computing resources. Results provide detailed views of how nature might correct for 'mistakes' in binding orientation.
Click here to visit the Chong group’s website.
Coalson group
Theory of Chemical Dynamics
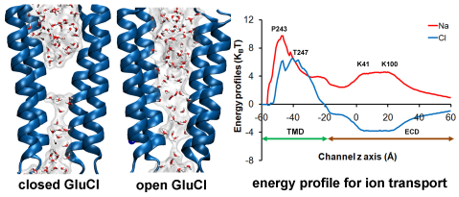 Research in the Coalson group focuses on the following topics: (1) Structure-function relations in ion channels and related biological nanopores, including ion permeation characteristics and gating mechanisms. (2) Structure-function relations in Nuclear Pore Complexes (NPCs), including mechanisms for transporting large cargos across the nuclear envelope. (3) Equilibrium and dynamical properties of polymer brushes. (4) Structural and dynamical features of Colloidal Crystal Arrays (CCAs). (5) Methods for efficient numerical calculation of protein conformational motion, with emphasis on internal rigid body decomposition.
Research in the Coalson group focuses on the following topics: (1) Structure-function relations in ion channels and related biological nanopores, including ion permeation characteristics and gating mechanisms. (2) Structure-function relations in Nuclear Pore Complexes (NPCs), including mechanisms for transporting large cargos across the nuclear envelope. (3) Equilibrium and dynamical properties of polymer brushes. (4) Structural and dynamical features of Colloidal Crystal Arrays (CCAs). (5) Methods for efficient numerical calculation of protein conformational motion, with emphasis on internal rigid body decomposition.
Click here to visit the Coalson group’s website.
Garrett-Roe group
Ultrafast Dynamics; Nonlinear Spectroscopy; Biophysics; Solvation Dynamics; Ion Channels; Ionic Liquids
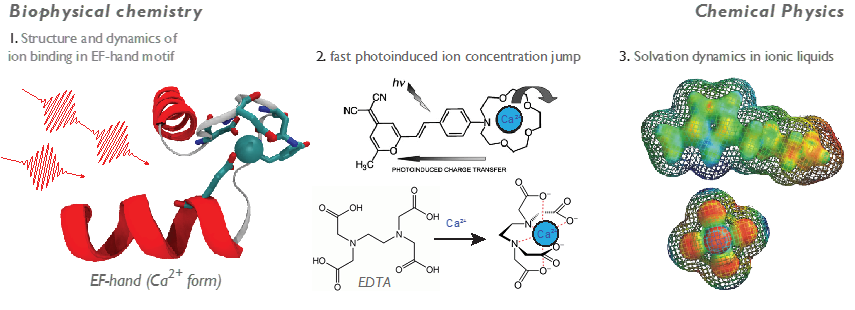
An urgent problem in protein science is to understand ion uptake and ion recognition (selectivity) by proteins and polypeptides. Why and how are these bio-‐nanostructures so exquisitely sensitive to particular ions? What are the elementary kinetic steps to ion uptake? In a separate field, that of green chemistry, there is another pressing problem to understand structural and dynamical heterogeneity in ionic liquids. Better understanding the chemical physics of these systems will aid the rational design of new solvents, which in turn will have industrial consequences like improved separation of CO2 from flue gas (carbon sequestration). There is an underlying connection between these biophysical and chemical physics questions – to really probe the mechanisms in operation, one must be able to separate static and dynamic heterogeneity. The nonlinear spectroscopies developed in the Garrett‐Roe lab can reveal structural dynamics on timescales spanning femtosecond–microseconds, making molecular movies which will answer these questions.
Click here to visit the Garrett-Roe group’s website.
Hutchison group
Materials Chemistry, Theoretical and Computational Chemistry, Advanced Functional Materials, Computational Materials Design, Inorganic Synthesis
 The Hutchison group focuses on combined simulation and experiments for rapidly designing high-efficiency organic electronics for solar cells, molecular piezoelectrics, and more. They have pioneered the "bottom-up" design of piezoelectric materials from single-molecule springs using both computational design and piezo-force microscopy (PFM) on single monolayers. In collaboration with the Lambrecht group, they have studied the use of hydrogen bonding for piezoelectric self-assembled smart materials and the rational design of mechanical energy harvesting devices. Similar approaches have been used to push the frontiers of organic solar cells. The group pioneered the use of evolutionary algorithms to rapidly find new high-efficiency targets with near-optimal electronic and optical properties using as few quantum calculations as possible. We also use high-performance Monte Carlo charge dynamics simulations in combination with Kelvin-probe microscopy (KPM) to understand the charge transport in the presence of defects and disorder in these organic semiconductors.
The Hutchison group focuses on combined simulation and experiments for rapidly designing high-efficiency organic electronics for solar cells, molecular piezoelectrics, and more. They have pioneered the "bottom-up" design of piezoelectric materials from single-molecule springs using both computational design and piezo-force microscopy (PFM) on single monolayers. In collaboration with the Lambrecht group, they have studied the use of hydrogen bonding for piezoelectric self-assembled smart materials and the rational design of mechanical energy harvesting devices. Similar approaches have been used to push the frontiers of organic solar cells. The group pioneered the use of evolutionary algorithms to rapidly find new high-efficiency targets with near-optimal electronic and optical properties using as few quantum calculations as possible. We also use high-performance Monte Carlo charge dynamics simulations in combination with Kelvin-probe microscopy (KPM) to understand the charge transport in the presence of defects and disorder in these organic semiconductors.
Click here to visit the Hutchison group’s website.
Jordan group
Theoretical and Computational Chemistry; Reactions on surfaces, properties of molecular hydrgen-bonded, Monte Carlo and molecular dynamics simulations; Electron-molecule interactions
 Professor Jordan's group is engaged in theoretical and experimental studies of the properties of molecules and clusters, of reaction at surfaces, of electron and proton localization and transfer in polyatomic molecules and water clusters. The Jordan group is developing new methods for treating long-range electron correlation effects, and using accurate electronic structure methods to develop improved force fields and model Hamiltonians for describing non-valence anions. We are also engaged in the use of quantum Monte Carlo approaches to electronic structure, and in the use of computational methods to address problems in the sustainable energy area.
Professor Jordan's group is engaged in theoretical and experimental studies of the properties of molecules and clusters, of reaction at surfaces, of electron and proton localization and transfer in polyatomic molecules and water clusters. The Jordan group is developing new methods for treating long-range electron correlation effects, and using accurate electronic structure methods to develop improved force fields and model Hamiltonians for describing non-valence anions. We are also engaged in the use of quantum Monte Carlo approaches to electronic structure, and in the use of computational methods to address problems in the sustainable energy area.
Click here to visit the Jordan group’s website.
Laaser group
Physics and Chemistry of Polymeric Materials
 Research in the Laaser group is at the interface between physical chemistry and polymeric materials, and focuses on understanding how changes in the molecular-scale interactions in polymer systems translate to changes in bulk materials properties. We utilize both controlled polymer design, in which polymers are designed to "turn on" or "turn off" the interactions of interest, and optical and spectroscopic techniques capable of probing various chromophores as reporters of the molecular environment and interactions. We are particularly interested in using these techniques to understand the properties of responsive polymers in order to inform application-specific polymer design. Current projects include work on the fundamental phase behavior and applications of bulk polyelectrolyte complexes and on the molecular-level dynamics of physically cross-linked polymers under deformation, among many others.
Research in the Laaser group is at the interface between physical chemistry and polymeric materials, and focuses on understanding how changes in the molecular-scale interactions in polymer systems translate to changes in bulk materials properties. We utilize both controlled polymer design, in which polymers are designed to "turn on" or "turn off" the interactions of interest, and optical and spectroscopic techniques capable of probing various chromophores as reporters of the molecular environment and interactions. We are particularly interested in using these techniques to understand the properties of responsive polymers in order to inform application-specific polymer design. Current projects include work on the fundamental phase behavior and applications of bulk polyelectrolyte complexes and on the molecular-level dynamics of physically cross-linked polymers under deformation, among many others.
Click here to visith the Laaser group's website.
H. Liu group
Physical and synthetic chemistries of nanomaterials including but not limited to DNA, graphene, carbon nanotubes, and colloidal nanocrystals
 Research in the H. Liu group is focused on the synthesis, characterization, and application of nanomaterials, in particular, graphene, DNA nanostructures, and semiconductor nanocrystals. Our graphene research is focused on understanding the surface properties of graphene and how they are affected by the environment. Our recent work in this area covers a wide range of topics, including wettability (Li, Z.; et al. Nature Mater. 2013), chemical reactivity (Surwade, S.; et al. J Phys. Chem. C, 2012), anti-corrosion properties (Zhou, F.; et al. ACS Nano 2013), and tribology of graphene (Kozbial, A.; et al. Thin Solid Films, 2013).
Research in the H. Liu group is focused on the synthesis, characterization, and application of nanomaterials, in particular, graphene, DNA nanostructures, and semiconductor nanocrystals. Our graphene research is focused on understanding the surface properties of graphene and how they are affected by the environment. Our recent work in this area covers a wide range of topics, including wettability (Li, Z.; et al. Nature Mater. 2013), chemical reactivity (Surwade, S.; et al. J Phys. Chem. C, 2012), anti-corrosion properties (Zhou, F.; et al. ACS Nano 2013), and tribology of graphene (Kozbial, A.; et al. Thin Solid Films, 2013).
Click here to visit the H. Liu group’s website.
Peng Liu group
Computational Organic Chemistry, Transition Metal-Catalyzed Reactions
 Professor Peng Liu’s group uses computational tools to study and predict reactivity and selectivity in organic reactions. Currently, we are focusing on transition metal-catalyzed carbon-hydrogen and carbon-carbon bond functionalization and olefin hydrofunctionalization reactions. We perform quantum mechanical calculations to explore the reaction mechanisms and various steric, electronic, and stereoelectronic effects to predict how perturbations of the catalyst structure, substituents, and solvent affect rate and selectivity. The theoretical insights are being incorporated to develop catalyst screening algorithms and software for rapid computational discovery of new catalysts.
Professor Peng Liu’s group uses computational tools to study and predict reactivity and selectivity in organic reactions. Currently, we are focusing on transition metal-catalyzed carbon-hydrogen and carbon-carbon bond functionalization and olefin hydrofunctionalization reactions. We perform quantum mechanical calculations to explore the reaction mechanisms and various steric, electronic, and stereoelectronic effects to predict how perturbations of the catalyst structure, substituents, and solvent affect rate and selectivity. The theoretical insights are being incorporated to develop catalyst screening algorithms and software for rapid computational discovery of new catalysts.
Click here to visit the P. Liu group’s website.
Millstone group
Inorganic and Materials Chemistry; Nanomaterials; Mechanochemistry; Surface and Colloid Chemistry
 Nanoscale materials are driving the discovery of physical and chemical phenomena, and providing the basis for cutting-edge technologies that range from low-cost solar cells to targeted gene therapies. At the heart of these advances are controlled chemical synthesis and the development of structure-property relationships. The Millstone group combines tools from molecular chemistry, materials science, and surface spectroscopy to build a detailed, bottom-up understanding of the processes and properties that drive the behavior of colloidal nanoparticles. Using this information we prepare new structures that incorporate optical, electronic, mechanical, and self-assembly demands into a single nanoparticle architecture. This work will not only provide significant insight into nanoscale reactions and particle properties, but also provide new particle platforms, and dramatically accelerate the translation of nanoparticles into society-shaping technologies.
Nanoscale materials are driving the discovery of physical and chemical phenomena, and providing the basis for cutting-edge technologies that range from low-cost solar cells to targeted gene therapies. At the heart of these advances are controlled chemical synthesis and the development of structure-property relationships. The Millstone group combines tools from molecular chemistry, materials science, and surface spectroscopy to build a detailed, bottom-up understanding of the processes and properties that drive the behavior of colloidal nanoparticles. Using this information we prepare new structures that incorporate optical, electronic, mechanical, and self-assembly demands into a single nanoparticle architecture. This work will not only provide significant insight into nanoscale reactions and particle properties, but also provide new particle platforms, and dramatically accelerate the translation of nanoparticles into society-shaping technologies.
Click here to visit the Millstone group’s website.
Saxena group
Analytical, Biophysical, and Physical Chemistry
 We develop pulsed electron spin resonance methods and their application to otherwise inaccessible problems in biophysics and materials sciences. The coupling of electron spin angular momentum to its environment—as revealed by the ESR spectrum—provides rich information about the electronic, structural and dynamical properties of the molecule. We create methods that measure the precise distance between two units in a protein, in order to determine their folding patterns and conformational dynamics. These ESR Spectroscopic Rulers— based on multiple quantum coherences and double resonance experiments—are unique in that they resolve distances in the 1 - 7 nm lengthscale even on bulk amorphous materials. Much of this work is based on the use of first-principles theory to develop new experimental protocols and to analyze experimental results.
We develop pulsed electron spin resonance methods and their application to otherwise inaccessible problems in biophysics and materials sciences. The coupling of electron spin angular momentum to its environment—as revealed by the ESR spectrum—provides rich information about the electronic, structural and dynamical properties of the molecule. We create methods that measure the precise distance between two units in a protein, in order to determine their folding patterns and conformational dynamics. These ESR Spectroscopic Rulers— based on multiple quantum coherences and double resonance experiments—are unique in that they resolve distances in the 1 - 7 nm lengthscale even on bulk amorphous materials. Much of this work is based on the use of first-principles theory to develop new experimental protocols and to analyze experimental results.
Our group continues to develop applications of these spectroscopic rulers that range from capturing the essence of structural changes - such as misfolding - in proteins, to measuring the atomic-level details of ion-permeation in a ligand gated ion-channel. We invite you to visit our group website and to contact us to explore the diversity of research projects currently underway in our group.
Click here to visit the Saxena group's website.
Waldeck group
The Waldeck group is investigating the Chiral Induced Spin Selectivity (CISS) effect and its manifestations in spintronics, chemistry, and biology.
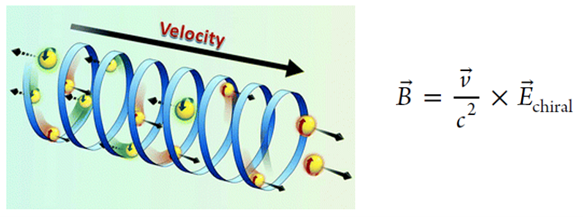
CISS dates from 1999 (K.Ray et al. Science 283, 814), in which Waldeck and Naaman reported that the transmission yield of spin-polarized electrons through chiral Langmuir-Blodgett films depends on the handedness of the molecular films. Since that time, CISS has been shown to be manifest in electron tunneling processes, electron transfer reactions, and in ferromagnetic surface/molecule exchange interactions. The Waldeck group is exploring fundamental aspects of CISS in supramolecular assemblies of chiral molecules and chiral materials and is using CISS to improve the efficiency and selectivity of electrochemical reactions and optoelectronic materials.
See the Waldeck group website for more information:
The CISS Effect - THE WALDECK LAB https://www.waldecklab.com/the-ciss-effect.html














